“This research demonstrates that if Australian adults use grain fibre to increase their intake of dietary fibre to target intake levels for chronic disease risk reduction (28g for women, 38g for men):
• The potential healthcare expenditure savings would be approximately $1 billion for CVD and over $285 million for T2D in 2015–16. The savings for CVD would represent approximately 0.6% of total Australian health expenditure and savings for T2D would be around 0.2% of health expenditure.
• The potential productivity cost savings were estimated to be approximately $600 million for CVD and $1.4 billion for T2D. The savings for CVD represent approximately 0.04% of gross domestic product (GDP) and for T2D, approximately 0.08% of GDP.
The total combined economic savings could potentially reach $3.3 billion.”
Zoe Harcombe looks into the evidence base here and finds it lacking; however as her article requires a subscription and she asked me to look at the evidence to try to make sense of some convoluted manipulations, I'm going to discuss some extra aspects of the main type 2 diabetes study used, the EPIC-InterAct study and its meta-analysis.[1]
This paper presents the results from a large multicentre epidemiological study from the EPIC cohorts, followed by what is supposed to be a meta-analysis of prospective epidemiological studies (more on that later).The EPIC-InterAct data is the "news" here, and it doesn't support the hypothesis that cereal fibre prevents type 2 diabetes when adjusted for age and sex, or for "lifestyle, diet and BMI" but does in the purely lifestyle and diet adjusted models.
However, if we look at the forest plot, we can see that the associations are all over the place, and there are many countries without a protective association, including France with a non-significant HR of 1.72. Another outlier is the UK with an HR of 0.74 (NS).
What are the differences in these populations? The French cohorts, for some reason, are all 100% female, which isn't the case for any other country. And the largest of the two UK cohorts is EPIC-Oxford.[2]
"The majority of participants recruited by the EPIC Oxford (UK) centre consisted of vegetarian and “health conscious” volunteers from England, Wales, Scotland, and Northern Ireland"
So these health conscious volunteers are probably being compared with people with lower fibre intakes in the less health-conscious UK cohort.
Sweden always interests me because one of their two cohorts is the Malmo Diet and Cancer Study, which uses a 7-day food diary for all subjects, and in Sweden the HR is a more reasonable 0.96. So less confounding by conscientiousness and more accurate diet recall tends to minimize the cereal fibre and type 2 diabetes association. If cereal fibre prevented type 2 diabetes in any important way, it should show up in most of these populations, not just a few.
The InterAct authors then follow up this rather inconsistent evidence by including it in a meta-analysis of all other prospective cohort studies on the question. This is an example of advocacy epidemiology - our study didn't convincingly support our belief, so we'll incorporate it into the totality of less relevant evidence that does. Presumably an intention of InterAct is to inform European dietary recommendations, for which the European evidence is most relevant, and for which it seems to say that the effect of cereal fibre on type 2 diabetes risk depends a great deal on what country you're a citizen of, so cannot be guaranteed either way.
Lo and behold the larger meta produces the strongly supportive associations that informed the Australian Kellogg's paper.
But hang on.
At least one of the studies included, the Finnish Diabetes Prevention Study, isn't a prospective cohort study at all - it's a mixed intervention of diet and exercise advice vs no intervention.
This simply should not have been included. It's a small study so shouldn't have biased the result too much if at all, but it doesn't inspire confidence that the selection of studies for inclusion was as rigorous as it should have been. (It's discussed and excluded from an analysis in a supplementary paper, so this isn't an error).
There are 3 Australian studies, two small ones with protective associations and a large one, Hodge 2004, with none.[3] However Hodge 2004 finds that white bread is associated with type 2 diabetes, and that lower GI carbs, including sugar, aren't. White bread in Australia is so bad that even sugar looks good by comparison. Does it follow that putting a few grams of bran in white bread will improve it? Why not just say "avoid white bread"? Anyway, if fibre isn't associated with type 2 diabetes in a fairly large sample of Australians, who as Zoe Harcombe pointed out tend to have fairly high fibre intakes anyway by OECD standards, why do Aussies need to take their lead from the USA?
Because most of the weighty studies in favour of fibre for type 2 diabetes prevention in the meta-analysis come from the USA. There are two important facts about the USA - low fibre intakes are lower than they are anywhere else (so basically a high intake of deep fried food, white breads, and soda in these groups), and conscientiousness is an identifiable confounding factor in many populations. For example, the Nurses' Health Study and Health Professionals' Follow-up Study; here we have the populations not only given the most advice about fibre being healthy, but also given the job of passing it on to the other US populations. In other words, grain fibre - like red meat - is one of the signifiers separating conscientious Americans from other Americans (it's harder to eat fruit and vege without their fibre, and vege in the US includes fried potatoes). It's almost a class distinction.
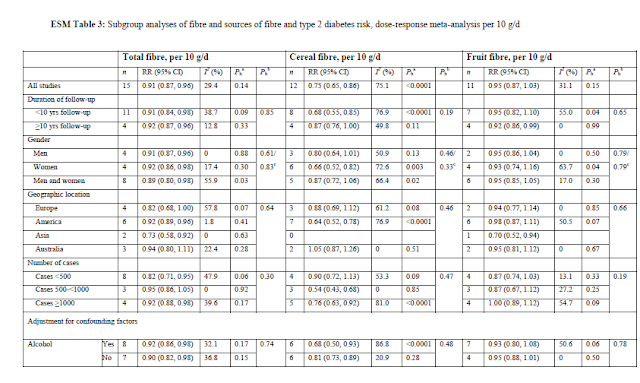
Look at this subgroup analysis of the "dose response" (ESM Table 3) - it's ALL about the USA. Zoe and I couldn't get our heads around how this "dose-response" was calculated from such diverse studies that all had differing groupings and cut-offs, but even taken at face value, why would an Australian government claim it showed any health savings for Australians?
If we compare diabetes prevalence between countries with mean fibre intakes, the USA with its abnormally low fibre intake for a high-cereal society fits, but other countries don't.
This paper presents the results from a large multicentre epidemiological study from the EPIC cohorts, followed by what is supposed to be a meta-analysis of prospective epidemiological studies (more on that later).The EPIC-InterAct data is the "news" here, and it doesn't support the hypothesis that cereal fibre prevents type 2 diabetes when adjusted for age and sex, or for "lifestyle, diet and BMI" but does in the purely lifestyle and diet adjusted models.
However, if we look at the forest plot, we can see that the associations are all over the place, and there are many countries without a protective association, including France with a non-significant HR of 1.72. Another outlier is the UK with an HR of 0.74 (NS).
What are the differences in these populations? The French cohorts, for some reason, are all 100% female, which isn't the case for any other country. And the largest of the two UK cohorts is EPIC-Oxford.[2]
"The majority of participants recruited by the EPIC Oxford (UK) centre consisted of vegetarian and “health conscious” volunteers from England, Wales, Scotland, and Northern Ireland"
So these health conscious volunteers are probably being compared with people with lower fibre intakes in the less health-conscious UK cohort.
Sweden always interests me because one of their two cohorts is the Malmo Diet and Cancer Study, which uses a 7-day food diary for all subjects, and in Sweden the HR is a more reasonable 0.96. So less confounding by conscientiousness and more accurate diet recall tends to minimize the cereal fibre and type 2 diabetes association. If cereal fibre prevented type 2 diabetes in any important way, it should show up in most of these populations, not just a few.
The InterAct authors then follow up this rather inconsistent evidence by including it in a meta-analysis of all other prospective cohort studies on the question. This is an example of advocacy epidemiology - our study didn't convincingly support our belief, so we'll incorporate it into the totality of less relevant evidence that does. Presumably an intention of InterAct is to inform European dietary recommendations, for which the European evidence is most relevant, and for which it seems to say that the effect of cereal fibre on type 2 diabetes risk depends a great deal on what country you're a citizen of, so cannot be guaranteed either way.
Lo and behold the larger meta produces the strongly supportive associations that informed the Australian Kellogg's paper.
But hang on.
At least one of the studies included, the Finnish Diabetes Prevention Study, isn't a prospective cohort study at all - it's a mixed intervention of diet and exercise advice vs no intervention.
This simply should not have been included. It's a small study so shouldn't have biased the result too much if at all, but it doesn't inspire confidence that the selection of studies for inclusion was as rigorous as it should have been. (It's discussed and excluded from an analysis in a supplementary paper, so this isn't an error).
There are 3 Australian studies, two small ones with protective associations and a large one, Hodge 2004, with none.[3] However Hodge 2004 finds that white bread is associated with type 2 diabetes, and that lower GI carbs, including sugar, aren't. White bread in Australia is so bad that even sugar looks good by comparison. Does it follow that putting a few grams of bran in white bread will improve it? Why not just say "avoid white bread"? Anyway, if fibre isn't associated with type 2 diabetes in a fairly large sample of Australians, who as Zoe Harcombe pointed out tend to have fairly high fibre intakes anyway by OECD standards, why do Aussies need to take their lead from the USA?
Because most of the weighty studies in favour of fibre for type 2 diabetes prevention in the meta-analysis come from the USA. There are two important facts about the USA - low fibre intakes are lower than they are anywhere else (so basically a high intake of deep fried food, white breads, and soda in these groups), and conscientiousness is an identifiable confounding factor in many populations. For example, the Nurses' Health Study and Health Professionals' Follow-up Study; here we have the populations not only given the most advice about fibre being healthy, but also given the job of passing it on to the other US populations. In other words, grain fibre - like red meat - is one of the signifiers separating conscientious Americans from other Americans (it's harder to eat fruit and vege without their fibre, and vege in the US includes fried potatoes). It's almost a class distinction.

Look at this subgroup analysis of the "dose response" (ESM Table 3) - it's ALL about the USA. Zoe and I couldn't get our heads around how this "dose-response" was calculated from such diverse studies that all had differing groupings and cut-offs, but even taken at face value, why would an Australian government claim it showed any health savings for Australians?
If we compare diabetes prevalence between countries with mean fibre intakes, the USA with its abnormally low fibre intake for a high-cereal society fits, but other countries don't.
"The mean±SD fibre intake in the subcohort was 22.9± 6.2 g/day (ranging from 19.9 g/day in Sweden to 25.2 g/day in Denmark; data not shown)."
Germany (7.40%) and Denmark (7.20%) have higher diabetes prevalence rates than Sweden (4.70%). USA's mean total fibre intake is 16.1 g/day and diabetes prevalence is 10.80%.
Germany and Denmark were the other populations in EPIC-InterAct where fibre had some protective association with type 2 diabetes. I'm not sure about T2D, but the amount of wholegrain associated with a modest degree of ischaemic CVD protection in Malmo was only 2.5 servings a day, and there was a protective association between fibre (mostly cereal) and saturated fat (a high SFA, high fibre diet was the most protective combination).[4,5]
We are probably seeing - in all these studies - a protective effect of eating high quality food with a minimal human interference factor (dairy and ryebread in Scandinavia) and belonging to the social class that is more likely to do this. With regard to iCVD bran may be a "failsafe" source of silicon, needed for vascular resilience and repair (you can get silicon from other foods and water, but bran in a staple food would guarantee it was present in the diet), but it is hard to see how this applies to T2DM. If a microbiotal mechanism was strongly involved, presumably other types of fibre would be more protective than they are. I come back to what whole grains are not. They are not white bread, and what is white bread? It used to be made with alloxan, a chemical used to produce experimental diabetes in animals. Today it contains
INGREDIENTS: White Bread (Enriched Wheat Flour (Flour, Malted Barley Flour, Niacin, Iron(Ferrous Sulfate, Reduced Iron), Thiamine Mononitrate, Riboflavin, Folic Acid), Water, Yeast, Salt, Soybean Oil, Sugar, Malt, Dough Conditioners(Ascorbic Acid, Calcium Sulfate, Sodium Stearoyl Lactylate),Calcium Propionate(To Retard Mold Growth))
Germany and Denmark were the other populations in EPIC-InterAct where fibre had some protective association with type 2 diabetes. I'm not sure about T2D, but the amount of wholegrain associated with a modest degree of ischaemic CVD protection in Malmo was only 2.5 servings a day, and there was a protective association between fibre (mostly cereal) and saturated fat (a high SFA, high fibre diet was the most protective combination).[4,5]
We are probably seeing - in all these studies - a protective effect of eating high quality food with a minimal human interference factor (dairy and ryebread in Scandinavia) and belonging to the social class that is more likely to do this. With regard to iCVD bran may be a "failsafe" source of silicon, needed for vascular resilience and repair (you can get silicon from other foods and water, but bran in a staple food would guarantee it was present in the diet), but it is hard to see how this applies to T2DM. If a microbiotal mechanism was strongly involved, presumably other types of fibre would be more protective than they are. I come back to what whole grains are not. They are not white bread, and what is white bread? It used to be made with alloxan, a chemical used to produce experimental diabetes in animals. Today it contains
INGREDIENTS: White Bread (Enriched Wheat Flour (Flour, Malted Barley Flour, Niacin, Iron(Ferrous Sulfate, Reduced Iron), Thiamine Mononitrate, Riboflavin, Folic Acid), Water, Yeast, Salt, Soybean Oil, Sugar, Malt, Dough Conditioners(Ascorbic Acid, Calcium Sulfate, Sodium Stearoyl Lactylate),Calcium Propionate(To Retard Mold Growth))
ALLERGENS: Wheat, Soybeans, Gluten
This Harvard-supplied list is incomplete - NZ white bread contains soy protein; Tip Top's omega 3 loaf supplies;
This Harvard-supplied list is incomplete - NZ white bread contains soy protein; Tip Top's omega 3 loaf supplies;
Wheat Flour, Water, Soy Fibre, Baker's Yeast, Wheat Gluten, Vinegar, Iodised Salt, Vegetable Gum (412), Soy Flour, Canola Oil, Tuna Oil (0.05%) (Contains Fish, Soy), Milk Protein (Sodium Caseinate), Emulsifiers (481, 471, 472e), Vitamins (Thiamin, Folate, Vitamin E, Niacin, Vitamin B6), Minerals (Iron, Zinc).
Woah - the fibre isn't even grain fibre, and there are 3 emulsifiers. Emulsifiers are experimentally linked to obesity and diabetes.
So it's hard to rule out that whole grains just tend to replace a cause of type 2 diabetes in some societies.
So, if you're an American eating a lot of starch from fibre-free foods, replacing these with whole grain foods (which for one thing won't be cooked in oil) should decrease your risk of type 2 diabetes. Replacing them with anything closer to nature will likely have the same effect.
If you're a European, Brexit will be bad news, because you won't be able to move to the one place where fibre is really protective, but you can still go to Sweden and enjoy the best of both worlds.
If you're an Australian, ask yourself, do I eat like the average American who isn't health conscious? If the answer is yes, then sprinkling a bit of bran on your food won't save you. But if you avoid bread or cereals altogether, are you at extra risk of type 2 diabetes? That's what we really want to know, and what none of the epidemiology can tell us at all, unless it's the risk marker epidemiology, which as far as I know says low TG/HDL, high LDL, low HbA1c = lowest T2DM risk.
References
[1] The InterAct Consortium. Dietary fibre and incidence of type 2 diabetes in eight European countries: the EPIC-InterAct Study and a meta-analysis of prospective studies. Diabetologia. 2015;58(7):1394-1408. doi:10.1007/s00125-015-3585-9.ghbhttps://www.ncbi.nlm.nih.gov/pmc/articles/PMC4472947/
[2] The InterAct Consortium, Langenberg C, Sharp S, et al. The InterAct Project: An Examination of the Interaction of Genetic and Lifestyle Factors on the Incidence of Type 2 Diabetes in the EPIC Study. Diabetologia. 2011;54(9):2272-2282. doi:10.1007/s00125-011-2182-9.
If you're a European, Brexit will be bad news, because you won't be able to move to the one place where fibre is really protective, but you can still go to Sweden and enjoy the best of both worlds.
If you're an Australian, ask yourself, do I eat like the average American who isn't health conscious? If the answer is yes, then sprinkling a bit of bran on your food won't save you. But if you avoid bread or cereals altogether, are you at extra risk of type 2 diabetes? That's what we really want to know, and what none of the epidemiology can tell us at all, unless it's the risk marker epidemiology, which as far as I know says low TG/HDL, high LDL, low HbA1c = lowest T2DM risk.
References
[1] The InterAct Consortium. Dietary fibre and incidence of type 2 diabetes in eight European countries: the EPIC-InterAct Study and a meta-analysis of prospective studies. Diabetologia. 2015;58(7):1394-1408. doi:10.1007/s00125-015-3585-9.ghbhttps://www.ncbi.nlm.nih.gov/pmc/articles/PMC4472947/
[2] The InterAct Consortium, Langenberg C, Sharp S, et al. The InterAct Project: An Examination of the Interaction of Genetic and Lifestyle Factors on the Incidence of Type 2 Diabetes in the EPIC Study. Diabetologia. 2011;54(9):2272-2282. doi:10.1007/s00125-011-2182-9.
[3] Hodge AM, English DR, O'Dea K, Giles GG. Glycemic index and dietary fiber and the risk of type 2 diabetes. Diabetes Care. 2004;27:2701–2706. doi: 10.2337/diacare.27.11.2701
[4] Wallström P, Sonestedt E, Hlebowicz J, et al. Dietary Fiber and Saturated Fat Intake Associations with Cardiovascular Disease Differ by Sex in the Malmö Diet and Cancer Cohort: A Prospective Study. Obukhov AG, ed. PLoS ONE. 2012;7(2):e31637. doi:10.1371/journal.pone.0031637.
[5] Sonestedt E, Hellstrand S, Schulz C-A, et al. The Association between Carbohydrate-Rich Foods and Risk of Cardiovascular Disease Is Not Modified by Genetic Susceptibility to Dyslipidemia as Determined by 80 Validated Variants. Müller M, ed. PLoS ONE. 2015;10(4):e0126104. doi:10.1371/journal.pone.0126104.
[5] Sonestedt E, Hellstrand S, Schulz C-A, et al. The Association between Carbohydrate-Rich Foods and Risk of Cardiovascular Disease Is Not Modified by Genetic Susceptibility to Dyslipidemia as Determined by 80 Validated Variants. Müller M, ed. PLoS ONE. 2015;10(4):e0126104. doi:10.1371/journal.pone.0126104.


